
公共卫生学术热点追踪
Nat Genet | 林希虹团队发展全基因组测序数据罕见变异荟萃分析方法——MetaSTAAR
近年来,随着高通量测序技术的发展,全基因组测序成本大幅下降,越来越多的大规模全基因组测序研究 (whole genome sequencing, WGS) 正在迅速开展,例如50万人的英国生物样本库(UK Biobank)、美国国家心肺血液研究所(National Heart Lung and Blood Institute, NHLBI)20万人的精准化医学研究计划(Trans-Omics Precision Medicine Program, TOPMed)和美国国家人类基因组研究所(National Human Genome Research Institute)的35万人的基因组测序计划(Genome Sequencing Program, GSP)。荟萃分析通过共享汇总统计数据,为整合这些大型测序研究中的基因组数据并保护个体数据隐私提供了有效解决方案。然而大规模全基因组测序研究的荟萃分析受到海量遗传变异的挑战。目前已经进行了上百万个全基因组的测序,发现了近十亿个变异位点,其中罕见变异占比率超过99%[1]。这些变异的汇总统计数据,包括得分统计量(score statistics)信息和协方差(连锁不平衡,linkage disequilibrium, LD)矩阵信息,需要存储并共享。尽管目前已经有方法可以对罕见变异进行荟萃分析[2,3],但是这些方法需要海量空间来存储汇总统计数据,无法扩展到现有的大规模测序研究。同时,现有的方法无法控制人群结构和家系结构的混杂影响,也不能通过整合多组学功能注释数据提高分析的检验功效。因此,亟需针对大规模全基因组测序数据的罕见变异荟萃分析方法,以实现基因组大数据的共享,找到导致疾病的遗传变异位点,发现人类疾病和表型的遗传构架,并开发新的药物标靶。
2022年12月23日,哈佛大学陈曾熙公共卫生学院林希虹院士团队在Nature Genetics杂志上发表了题为Powerful, scalable and resource-efficient meta-analysis of rare variant associations in large whole genome sequencing studies 的研究论文,发展了大规模全基因组测序数据罕见变异荟萃分析方法MetaSTAAR,实现了全基因组汇总统计数据的高效存储和功能知情的荟萃分析。
MetaSTAAR方法发展了一种基于稀疏矩阵的汇总统计数据存储方法,突破了测序数据罕见变异汇总统计数据的存储瓶颈,实现了大规模全基因组测序数据的功能知情的罕见变异荟萃分析(图1)。首先, MetaSTAAR利用测序数据中罕见变异基因型的稀疏性,发展了汇总统计数据存储的新方法,比同类方法节省了数百倍的存储空间。其次,MetaSTAAR可以控制人群结构和家系结构的混杂影响,适用于包括连续型和离散型在内多种类型的表型数据。最后,MetaSTAAR通过STAAR框架有效地整合了罕见变异多方面的生物功能信息[4],达到了提高罕见变异关联性荟萃分析检验功效的目的。这一研究为全基因组测序数据荟萃分析提供了高效和高性能的分析方法,突破了大规模全基因组测序研究汇总统计数据存储瓶颈和荟萃分析运算瓶颈,填补了大规模全基因组测序数据中罕见变异关联分析方法的重要空白。
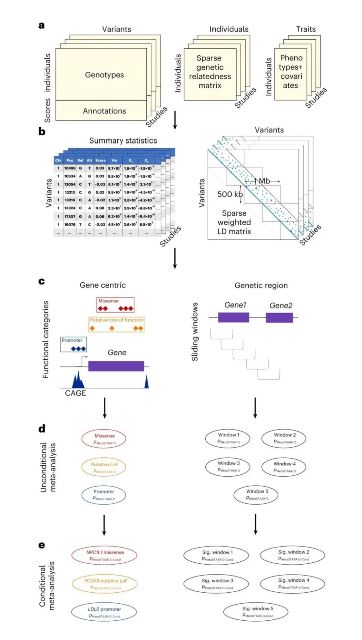
图1 MetaSTAAR荟萃分析流程。(1)每项研究的输入数据,包括基因型和表型数据、应用FAVORannotator工具得到的功能注释数据、稀疏遗传相关性矩阵。(2)应用MetaSTAARWorker生成各项研究的全基因组汇总统计数据。(3)全基因组荟萃分析,包括常见变异的单体荟萃分析和罕见变异的集合荟萃分析。集合荟萃分析中的分析单位包括功能类别集和滑动窗口。(4)荟萃分析结果汇总。(5)条件荟萃分析。
研究团队将MetaSTAAR应用到TOPMed中的全基因组测序数据中,对14项研究的30,138个样本中的4种脂质性状进行了荟萃分析,发现了与脂质性状显著相关的罕见变异。研究团队进一步应用MetaSTAAR方法对约200,000个样本的UK Biobank全外显子组测序数据和30,138个样本的TOPMed全基因组测序数据进行了罕见变异荟萃分析。所有的荟萃分析均使用高效的基于稀疏矩阵的新方法存储了汇总统计数据,并且可以快速地完成了常见变异和罕见变异荟萃分析。这些结果表明,MetaSTAAR使用高效的稀疏矩阵方法存储了全基因组测序数据的汇总统计数据,在大规模队列中实现功能知情的罕见变异关联性分析的同时,保护研究参与者的基因组数据隐私,实现了基因组大数据的共享和整合。
综上所述,林希虹院士团队开发的MetaSTAAR方法实现了全基因组测序数据高效和高性能的荟萃分析,为海量基因组大数据的共享和整合奠定了基础,有助于加快新药物标靶的发现和精准健康的研究。MetaSTAAR的R软件实现可以从网站https://github.com/xihaoli/MetaSTAAR下载。
哈佛大学教授、美国国家医学院林希虹院士和印第安纳大学医学院助理教授李子林为本文的共同通讯作者,林希虹院士课题组厉希豪博士后研究员为本文的第一作者。
(来源:BioArt)
参考文献:
1. Taliun, D. et al. Sequencing of 53,831 diverse genomes from the NHLBI TOPMed Program. Nature 590, 290-299 (2021).
2. Lee, S., Teslovich, Tanya M., Boehnke, M. & Lin, X. General Framework for Meta-analysis of Rare Variants in Sequencing Association Studies. The American Journal of Human Genetics 93, 42-53 (2013).
3. Liu, D.J. et al. Meta-analysis of gene-level tests for rare variant association. Nature Genetics 46, 200-204 (2014).
4. Li, X. et al. Dynamic incorporation of multiple in silico functional annotations empowers rare variant association analysis of large whole-genome sequencing studies at scale. Nature Genetics 52, 969-983 (2020).