
公共卫生学术热点追踪
Immunity|HIV潜伏感染的激活机制
抗逆转录病毒治疗(Antiretroviral therapy ART)能够有效阻断病毒复制,但HIV-1仍然潜伏在一小部分具有诱导性且具有复制能力的潜伏感染静止CD4+ T细胞中。这一病毒储存库最初是通过定量病毒产出实验进行表征的,在实验中,静止的CD4+ T细胞通过能引发多克隆T细胞激活的试剂(如植物血凝素以及抗-CD3和抗-CD28抗体的组合)被刺激。激活会引发转录环境的变化,包括NF-kB和NFAT转录因子向细胞核的转移,从而促进HIV-1基因表达。定量病毒产出实验显示了储存库极其缓慢的衰减速率,这使得必须长期依赖ART。中断ART会导致病毒快速反弹,可能是由于潜伏感染细胞的激活所致。预防这种反弹是HIV-1治愈研究的主要目标,但诱导体内潜伏HIV-1的刺激性质仍然不明确。一个假说认为,识别特异性抗原的事件是导致反弹的关键,但由于潜伏感染细胞的抗原特异性未知,这一假说的验证存在挑战。
管抗原刺激在病毒反弹中的作用尚不明确,但已有强有力的证据表明,抗原在潜伏病毒储存库的持久性中发挥关键作用,驱动了感染细胞的增殖。事实上,来自持续性病毒感染(如HIV1和CMV)的抗原反复暴露,可以促使带有特定抗原T细胞受体的感染CD4+T细胞发生大规模克隆扩增。这一过程或许可以解释,为什么在接受抗逆转录病毒治疗超过20年的人群中,潜伏感染细胞的频率会趋于稳定甚至增加。
近日,美国约翰霍普金斯大学的Francesco R. Simonetti 团队在Immunity上发表题为Cognate antigen engagement induces HIV-1 expression in latently infected CD4+ T cells from people on long-term antiretroviral therapy 的文章。该文章发现抗原的识别可驱动 HIV-1 基因在潜伏感染的 CD4+ T 细胞中表达,这表明抗原刺激可能在长期抗逆转录病毒治疗中残余病毒库的持久性和治疗中断后的病毒反弹中起关键作用。

作者首先从长期ART患者中分离抗原反应性CD4+ T细胞。使用自体树突状细胞(DCs)加载CMV裂解物或HIV-1 Gag肽抗原,刺激这些细胞。测定刺激后细胞内HIV-1 RNA水平。结果显示自体DC呈递的特异性抗原显著提高了HIV-1 RNA表达,表明抗原识别可诱导HIV-1转录。接下来作者通过流式细胞术分析抗原刺激后CD4+ T细胞的激活标志物(CD69和CD154)的表达。对扩增后的抗原反应性细胞使用单细胞极限稀释法,测定单个细胞中HIV-1 RNA拷贝数。抗原刺激显著激活T细胞,但只有极少数细胞产生高水平的HIV-1 RNA,这表明T细胞激活与HIV-1表达存在解耦。
作者又比较了多克隆刺激(PMA/I或抗-CD3/CD28抗体)与抗原特异性刺激(CMV或HIV-1 Gag抗原)诱导的HIV-1 RNA表达。使用RNA测序和qPCR分析转录变化。PMA/I和抗原特异性刺激均能诱导类似水平的HIV-1 RNA表达,两者作用的转录活性无显著差异。使用极限稀释法分离小群CD154+CD69+ T细胞,以单细胞分辨率测定每个细胞的HIV-1 RNA拷贝数。分析不同刺激条件下HIV-1 RNA阳性细胞的比例和转录水平。高水平HIV-1 RNA+细胞(>100 RNA拷贝)非常稀有,仅占所有HIV-1 RNA+细胞的3.8%,转录活动主要由少数细胞驱动。提取刺激后的HIV-1 RNA进行限制性稀释扩增和序列分析。评估不同刺激条件下的HIV-1 RNA序列特性和多样性。无论使用PMA/I还是特异性抗原刺激,诱导的HIV-1 RNA序列多为同一扩增克隆,表明这些刺激诱导了相同的感染细胞。
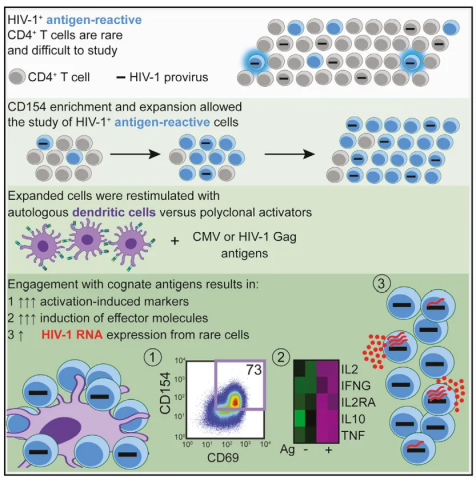
这些实验结果表明,抗原识别对HIV-1转录有直接影响,同时揭示了潜伏感染的激活机制和细胞异质性。
(来源:BioArtMED)
原文出处:Moskovljevic M, Dragoni F, Board NL, Wu F, Lai J, Zhang H, White JR, Hoh R, Lynn K, Tebas P, Mounzer K, Deeks SG, Montaner LJ, Siliciano JD, Siliciano RF, Simonetti FR. Cognate antigen engagement induces HIV-1 expression in latently infected CD4+ T cells from people on long-term antiretroviral therapy. Immunity. 2024 Dec 10;57(12):2928-2944.e6. doi: 10.1016/j.immuni.2024.11.002. Epub 2024 Nov 28. PMID: 39612916.